Articles
- Page Path
- HOME > J Powder Mater > Volume 32(3); 2025 > Article
-
Research Article
- Thermodynamic and Electronic Descriptor-Driven Machine Learning for Phase Prediction in High-Entropy Alloys: Experimental Validation
- Nguyen Lam Khoa, Nguyen Duy Khanh, Hoang Thi Ngoc Quyen, Nguyen Thi Hoang Oanh, Le Hong Thang, Nguyen Hoa Khiem, Nguyen Hoang Viet*
-
Journal of Powder Materials 2025;32(3):191-201.
DOI: https://doi.org/10.4150/jpm.2025.00143
Published online: June 30, 2025
Faculty of Materials Engineering, School of Materials Science and Engineering, Hanoi University of Science and Technology, Hanoi 100000, Vietnam
- *Corresponding author: Nguyen Hoang Viet E-mail: viet.nguyenhoang@hust.edu.vn
© The Korean Powder Metallurgy & Materials Institute
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 3,942 Views
- 109 Download
- 5 Crossref
Abstract
- High-entropy alloys (HEAs) exhibit complex phase formation behavior, challenging conventional predictive methods. This study presents a machine learning (ML) framework for phase prediction in HEAs, using a curated dataset of 648 experimentally characterized compositions and features derived from thermodynamic and electronic descriptors. Three classifiers—random forest, gradient boosting, and CatBoost—were trained and validated through cross-validation and testing. Gradient boosting achieved the highest accuracy, and valence electron concentration (VEC), atomic size mismatch (δ), and enthalpy of mixing (ΔHmix) were identified as the most influential features. The model predictions were experimentally verified using a non-equiatomic Al30Cu17.5Fe17.5Cr17.5Mn17.5 alloy and the equiatomic Cantor alloy (CoCrFeMnNi), both of which showed strong agreement with predicted phase structures. The results demonstrate that combining physically informed feature engineering with ML enables accurate and generalizable phase prediction, supporting accelerated HEA design.
- This integrated workflow combines descriptor engineering and experimental validation to develop robust machine learning models, offering a powerful tool for guiding the design of high-entropy alloys.
Graphical abstract
- High-entropy alloys (HEAs), also known as multi-principal element alloys, are a transformative class of metallic materials characterized by the presence of five or more principal elements in near-equimolar or significant proportions (typically 5 to 35 at.%) [1-16]. Their definition has evolved significantly in recent years. While the original definition emphasizes element count, contemporary research increasingly recognizes configurational entropy (ΔSmix ≥ 1.5R) as a more robust criterion, accommodating both non-equiatomic and quaternary systems [17]. Unlike conventional alloys, which are generally centered around a single principal element, HEAs derive their distinctive structural and functional characteristics from the synergistic interactions among multiple principal elements [18-20]. As a result, this compositional complexity gives rise to outstanding properties, including superior mechanical strength, excellent thermal stability, and enhanced corrosion resistance [19].
- The theoretical foundation of HEAs relies on four “core effects” [18]: the high-entropy effect, which stabilizes simple solid-solution phases (like FCC, BCC) over complex intermetallic compounds; the sluggish diffusion effect, which retards phase transformation kinetics [21]; the severe lattice-distortion effect, arising from atomic size differences, which causes lattice strain and enhances strength; and the cocktail effect, describing synergistic properties that are not merely the arithmetic averages of the constituent elements, but rather emergent properties arising from their interactions [22, 23].
- However, recent works have revisited these core effects, particularly questioning the universality of the sluggish diffusion and high-entropy stabilization mechanisms. For example, Hsu et al. [18] demonstrated that sluggish diffusion is not always observed, with differences noted between Mn-containing and Mn-free systems. Similarly, studies have pointed out that multi-phase HEAs can often exhibit multifunctional properties comparable to or even superior to those of single-phase systems [3, 19, 20], challenging the presumption that high configurational entropy alone guarantees solid-solution stability.
- Although HEAs hold great promise, accurately predicting and controlling their phase formation remain significant challenges, hindering the accelerated discovery and optimization of advanced materials. Phase formation in HEAs is governed by a delicate interplay of thermodynamic and kinetic factors, including the entropy and enthalpy of mixing (ΔSmix, ΔHmix) [24, 25], atomic size mismatch (δ), valence electron concentration (VEC), and electronegativity difference (Δχ) [26]. Several empirical and semi-empirical criteria have been proposed to guide phase prediction (e.g., extensions of Hume-Rothery rules, the Ω parameter, and VEC thresholds); however, accurate and generalizable prediction across the vast compositional space of HEAs remains difficult. Experimental exploration alone is often prohibitively costly and time-consuming, highlighting the need for data-driven predictive frameworks.
- In this context, previous studies such as Singh et al. [27] have applied machine learning to predict phases and design new HEA compositions. However, these efforts often rely on datasets with mixed-quality entries and less consistent processing histories, which may limit predictive robustness. In contrast, the present study introduces a machine learning framework trained on a rigorously curated experimental dataset and emphasizes physically meaningful descriptors tied directly to practical synthesis conditions. Furthermore, this study provides dual validation—computational and experimental—on both equiatomic and non-equiatomic systems, highlighting both predictive accuracy and practical feasibility.
- Recently, alloy design strategies have expanded beyond the strict equiatomic constraint towards non-equiatomic HEAs. This approach acknowledges that the stable solid solutions do not necessarily require equiatomic ratios, as the configurational entropy curve can be relatively flat near the equiatomic point, allowing a range of other compositions to possess similar entropy values and phase stability. This offers greater flexibility in alloy design for achieving desired properties and overcomes limitations encountered in some early equiatomic systems [28].
- Advances in machine learning (ML) have opened new avenues for modelling the complex, non-linear relationships between composition and phase stability in multi-component systems [29-31]. ML holds the potential to significantly accelerate the materials design and discovery process [32]. However, the predictive success of ML approaches critically depends on the quality of the input dataset, the physical relevance of the engineered features, and the ability to generalize beyond the training data. Moreover, explicitly validating ML predictions against experimentally synthesized and characterized HEA systems is crucial for building confidence in their practical applicability [33].
- In this study, we address these challenges by developing a machine learning-based framework for phase prediction in HEAs, grounded in a curated dataset of 648 experimentally characterized alloys. The input features were meticulously engineered based on thermodynamic and electronic descriptors and, importantly, were calculated from actual material preparation conditions using 10-gram powder batches precisely weighed according to target atomic fractions. Three state-of-the-art ML classifiers—Random Forest (RF), Gradient Boosting (GB), and CatBoost—were trained, hyperparameter-optimized, and evaluated for phase prediction. The model predictions were further validated through the experimental synthesis and characterization of two distinct systems: a novel non-equiatomic alloy (Al30Cu17.5Fe17.5Cr17.5Mn17.5) and the canonical equiatomic Cantor alloy (CoCrFeMnNi). This study demonstrates the predictive accuracy and robustness of the machine learning framework, providing a practical tool for accelerating the design of next-generation HEAs. Furthermore, this work highlights the critical role of physically meaningful feature engineering and experimental validation in enhancing the reliability of data-driven materials discovery.
1. Introduction
- This study employs a comprehensive methodology encompassing dataset construction, feature engineering grounded in physical metallurgy principles, machine learning model development and evaluation, and ultimately, experimental synthesis and validation. Detailed descriptions of each step are provided below to ensure reproducibility.
- 2.1. Dataset Compilation and Preprocessing
- The foundation of this work is a dataset comprising 648 distinct high-entropy alloy (HEA) compositions, curated by Professor Juliusz Dąbrowa (2019) from peer-reviewed experimental literature and further expanded with our own experiments. To promote consistency and minimize variability arising from different processing histories, the alloys included were predominantly synthesized via arc melting and evaluated in the as-cast condition. Any documented minor surface treatments were considered negligible for the primary purpose of phase classification. Each alloy entry was classified into one of several primary phase types commonly observed in HEA research: single-phase Body-Centered Cubic (BCC) or Face-Centered Cubic (FCC) solid solutions (SS), single-phase intermetallic compounds (IM), amorphous (AM) structures, or multi-phase (MP) mixtures such as BCC+FCC, BCC+IM, FCC+IM, or BCC+FCC+IM. Compositions resulting in complex, highly metastable, or poorly characterized phases were excluded from the dataset to enhance homogeneity and to focus on practically significant phase outcomes.
- 2.2 Feature Engineering and Descriptor Calculation
- To enable machine learning-based prediction of phase formation, compositional information was converted into quantitative descriptors reflecting fundamental thermodynamic, atomic, and electronic factors. Seven key features were engineered for each alloy:
- • Entropy of mixing (ΔSmix):
- Calculated by the formula [34]:
- where ci is the atomic fraction of element i and R is the universal gas constant (8.314 J•mol-1•K-1).
- • Enthalpy of mixing (ΔHmix):
- Determined using Miedema’s semi-empirical model [35], adapted for multi-component systems [24, 25, 35]:
- where Ωij is the binary mixing enthalpy between elements i and j.
- • Atomic size mismatch (δ):
- This parameter quantifies the degree of atomic packing disorder and is computed as follows [36]:
- where ri is the atomic radius of element i and
- • Valence electron concentration (VEC):
- Weighted-average of the valence electrons per atom, guiding phase stability towards FCC or BCC structures [36].
- • Electronegativity difference (Δχ):
- Calculated by the formula [34]:
- where χi is the electronegativity of element i and
- • Mean melting temperature (Tm): Averaged over all constituent elements using their atomic fractions.
- • Ω parameter:
- A combined stability indicator is defined as [36]:
- which integrates thermodynamic stability and chemical disorder.
- All features were normalized either to a [0,1] range or standard-scaled to prevent variables with large numeric magnitudes from dominating the learning process.
- 2.3. Machine Learning Model Development
- Three distinct machine learning algorithms were selected and implemented for HEA phase prediction: Random Forest (RF), Gradient Boosting (GB), and CatBoost [37, 38]. RF utilizes bootstrap aggregation (bagging) to build an ensemble of de-correlated decision trees, enhancing robustness and generalization. GB and CatBoost employ sequential boosting techniques, where each tree learns from the errors of the preceding ones; CatBoost incorporates specific enhancements for handling categorical features (though not heavily utilized here) and regularization. These models were chosen for their demonstrated strong performance in materials science classification tasks. They were implemented using the Python-based scikit-learn and CatBoost libraries within a Google Colab environment.
- A systematic hyperparameter optimization was performed using grid search coupled with 5-fold cross-validation executed exclusively on the training set (88% of the total dataset, leaving 12% as a hold-out test set). Key hyperparameters tuned included the number of estimators (trees/iterations), maximum tree depth, minimum samples per leaf/split, learning rate, and regularization terms (e.g., l2_leaf_reg for CatBoost), specific to each algorithm’s requirements.
- 2.4. Model Training, Validation, and Feature Importance Analysis
- After hyperparameter optimization, the models achieving the highest cross-validation scores were trained on the full training dataset (88%) and subsequently evaluated on a hold-out test set (12%). Model performance was assessed using standard classification metrics: accuracy, precision, recall, and F1-score.
- To interpret the models and understand the driving factors behind phase prediction, a feature importance analysis was conducted. Model-specific techniques were used: mean decrease in Gini impurity for Random Forest, and permutation importance for Gradient Boosting and CatBoost. This analysis aimed to rank the input features based on their contribution to the models’ predictive decisions [39].
- 2.5. Experimental Synthesis and Validation
- To rigorously validate the predictive capability of the developed ML framework, two distinct HEA compositions were selected for experimental synthesis and characterization: the novel non-equiatomic Al30Cu17.5Fe17.5Cr17.5Mn17.5 alloy and the canonical equiatomic CoCrFeMnNi (Cantor) alloy. Our samples were synthesized via mechanical alloying using a planetary ball mill (AGO-2 type), employing tungsten carbide (WC) milling media at a ball-to-powder ratio of 20:1, a rotation speed of 500 rpm, with stearic acid as the process control agent, all conducted under an argon atmosphere. Elemental powders (purity > 99.9%, particle size 45–75 μm) were accurately weighed in 10-gram batches to achieve the target atomic ratios, following process parameters consistent with established protocols in previous studies [40-43]. Phase identification was performed using X-ray diffraction (XRD) with Cu-Kα radiation (λ = 1.5406 Å), a 2θ range of 20°–100°, and a step size of 0.02°. The resulting diffraction patterns were analysed using a Python-based tool, as described in [44].
2. Methodology
- This section presents the outcomes derived from the analysis of the curated dataset, the performance evaluation of the developed machine learning models, and the subsequent experimental validation, both designed to assess the framework’s predictive capabilities. The findings are interpreted within the context of established physical metallurgy principles governing phase formation in high-entropy alloys.
- 3.1. Exploratory Data Analysis and Feature Relevance
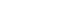
- Initial analysis of the curated dataset of 648 HEAs provided valuable insights into the relationships between the engineered features. Examination of the correlation heatmap (Fig. 1) revealed expected several physical trends. A strong positive correlation (0.92) was observed between the entropy of mixing (∆Smix) and the number of constituent elements, reflecting the fundamental definition of ideal configurational entropy. Furthermore, moderate negative correlations were identified between the valence electron concentration (VEC) and atomic size mismatch (δ) (-0.45), as well as between the enthalpy of mixing (ΔHmix) and δ (-0.68). These correlations suggest a complex interplay in which electronic structure, atomic packing efficiency, and chemical bonding affinity collectively influence phase stability within these multi-component systems.
- Analysis of the feature distributions (Fig. 2 and Table 1) further reinforced the physical relevance of the selected descriptors. VEC values were found to predominantly cluster between 6 and 8, aligning with established ranges governing FCC and BCC phase stability in HEAs. Similarly, the distribution of ΔHmix values centered near zero (mean = -8.335 kJ/mol), consistent with the thermodynamic competition between the formation of energetically favorable ordered intermetallic phases and entropically favored disordered solid solutions. These preliminary analyses confirmed the sound physical basis of the engineered features chosen for subsequent machine learning model development.
- 3.2. Machine Learning Model Performance and Feature Importance
- After conducting systematic hyperparameter optimization, we assessed the predictive performance of three models — Random Forest (RF), Gradient Boosting (GB), and CatBoost models — on the unseen hold-out test set (Table 2). All three models demonstrated high predictive accuracy, indicating their effectiveness in capturing the composition–phase relationships within the dataset (Fig. 3) [45]. The Gradient Boosting model exhibited slightly superior performance, achieving the highest cross–validation accuracy (88.5%, Table 2), and demonstrated strong generalization capabilities on the test set. These performance levels are competitive with those reported in other machine learning studies focused on HEA phase prediction. Consistent with observations in the literature, predicting phases involving intermetallic compounds was more challenging than predicting single-phase solid solutions.
- To explain the differences in hyperparameters, RandomForest employs hyperparameters like n_estimators, max_depth, min_samples_split, and min_samples_leaf to control the number of trees, tree depth, and minimum samples for splits or leaves, aligning with its bagging approach (independent tree training). In contrast, CatBoost and GradientBoosting, both based on gradient boosting, use similar parameters such as iterations/n_estimators and depth/max_depth, but include learning_rate to control the contribution of each tree during the boosting process. CatBoost also uses l2_leaf_reg for explicit regularization, while GradientBoosting relies on min_samples_leaf (like RandomForest) for implicit regularization. Additionally, RandomForest and GradientBoosting use min_samples_split to control node splitting, a parameter that CatBoost manages internally. These differences stem from their algorithms: RandomForest averages independent trees, whereas CatBoost and GradientBoosting train sequentially to correct errors, requiring additional parameters like learning_rate to balance performance and accuracy on the material dataset (predicting phases from features like ΔHmix, VEC).
- Feature importance analysis was conducted to elucidate the key factors driving the models’ predictions (Fig. 4). Across all three distinct algorithms, VEC consistently emerged as the most influential feature (~38–40% importance), underscoring its critical role in differentiating between FCC and BCC crystal structures. Atomic size mismatch (δ) (~14–21%) and enthalpy of mixing (ΔHmix) (~9–11%) were identified as the next most significant factors, primarily governing the propensity for forming disordered solid solutions versus ordered intermetallic or amorphous phases. The remaining features showed moderate importance, contributing to the overall phase stability landscape but playing secondary roles compared to VEC, δ, and ΔHmix in determining the primary phase structures within this dataset [36].
- 3.3. Model Application and Experimental Validation
- The predictive capability of the developed machine learning framework was assessed through its application to two distinct high-entropy alloy (HEA) systems: a novel non-equiatomic system, Al30Cu17.5Fe17.5Cr17.5Mn17.5, and the well-established equiatomic Cantor alloy, CoCrFeMnNi. For both alloys, thermodynamic and electronic descriptors including the entropy of mixing (ΔSmix), enthalpy of mixing (ΔHmix), valence electron concentration (VEC), atomic size mismatch (δ), electronegativity difference (Δχ), mean melting temperature (Tm), and the Ω parameter were calculated based on their compositions, and normalized prior to input into the trained models.
- In the case of Al30Cu17.5Fe17.5Cr17.5Mn17.5, all three machine learning classifiers—Random Forest, CatBoost, and Gradient Boosting—converged on a consistent prediction: that the dominant phase under standard synthesis conditions would be a single-phase BCC solid solution [36]. This prediction was experimentally verified by synthesizing the alloy through high-energy mechanical alloying (MA) for 3.5 hours, followed by heat-treatment at temperatures at 700 °C, as seen in Fig. 5. X-ray diffraction (XRD) analysis of the as-milled powders prior to heat-treatment revealed the presence of a dual-phase mixture comprising both FCC and BCC structures. The emergence of dual phases at this stage is attributed to competing thermodynamic forces—namely, a moderately negative ΔHmix and relatively high atomic size mismatch δ, combined with suboptimal entropy levels due to the non-equiatomic nature and dominant Al content [26]. These parameters position the alloy near the FCC-BCC phase boundary, and thus a kinetic pathway favoring multi-phase formation during MA is expected [36]. Upon heat-treatment, however, the material transformed into a well-defined single BCC phase, as evidenced by sharp diffraction peaks indicating improved crystallinity and phase purity at the temperature. This transformation from the initial dual-phase (FCC-BCC) mixture formed during MA is attributed to the high-temperature heat treatment effectively breaking down the metastable structure [28]. This strong agreement between predicted and experimentally observed stable phase supports the model’s effectiveness in anticipating thermodynamically favored structures for complex alloy systems [46].
- To further test the generality and robustness of the models, the equiatomic CoCrFeMnNi alloy was selected for validation. This composition is widely known for forming a stable single-phase FCC solid solution under a broad range of synthesis conditions, including non-equilibrium processing [16]. All models correctly predicted FCC as the primary phase, a result supported by computed descriptors: high configurational entropy (ΔSmix ≥ 1.5 R), moderate negative enthalpy of mixing (∼ − 2 kJ/mol), VEC ≈ 8.0 (strongly favoring FCC), atomic size mismatch δ < 5%, and an Ω value well within the stability regime of disordered FCC solid solutions.
- Experimentally, CoCrFeMnNi powders were subjected to mechanical alloying for durations between 1 and 20 hours, as shown in Fig. 6. Remarkably, even at the early stages of milling, XRD patterns showed only FCC peaks, with no evidence of BCC or intermetallic phases, demonstrating the inherent thermodynamic preference for FCC formation in this system. The immediate emergence of a stable FCC phase, even in the absence of any post-synthesis thermal treatment, underscores the role of high entropy and favorable elemental compatibility in suppressing phase competition during solid-state processing.
- The stark difference in phase evolution between the two systems can be understood through comparative analysis of their thermodynamic and kinetic descriptors. In Al30Cu17.5Fe17.5Cr17.5Mn17.5, the lower ΔSmix (relative to equiatomic HEAs), combined with strong atomic radius mismatch and a VEC (∼ 7.2) near the FCC-BCC transition zone, creates a compositional landscape prone to multiphase formation. The alloy requires thermal activation through sintering to homogenize the structure and enable the full transformation to the predicted BCC phase. In contrast, CoCrFeMnNi exhibits ideal HEA characteristics: equiatomic balance yields maximum configurational entropy, the VEC solidly resides in the FCC domain, and ΔHmix and δ remain within the threshold ranges that suppress ordered compound or BCC formation. As a result, this system achieves its predicted single FCC phase immediately after milling without requiring subsequent processing steps [47-49].
- These findings jointly validate the machine learning models not only in predicting the equilibrium phase of a complex non-equiatomic HEA after processing, but also in capturing phase behavior under non-equilibrium conditions for canonical HEA systems. The coherence between predicted and experimental phase outcomes across disparate compositions demonstrates the framework’s capacity to support accelerated HEA design and synthesis planning [50, 51].
3. Results and Discussion
- In this study, a machine learning framework was developed and validated for predicting phase formation in high-entropy alloys (HEAs). Using a curated dataset of 648 experimentally characterized alloys with engineered thermodynamic and electronic descriptors, three machine learning models—Random Forest, Gradient Boosting, and CatBoost—were trained and optimized. The models achieved high predictive accuracies, consistently identifying valence electron concentration (VEC), enthalpy of mixing (ΔHmix), and atomic size mismatch (δ) as the most critical factors governing phase stability.
- Experimental validation on two distinct HEA systems, a non-equiatomic Al30Cu17.5Fe17.5Cr17.5Mn17.5 alloy and the canonical CoCrFeMnNi alloy, demonstrated strong agreement between predicted and observed phase structures under different processing conditions. These results underscore the reliability and practical applicability of the developed ML framework in supporting accelerated alloy design.
- The successful integration of domain knowledge into feature engineering, along with systematic model training and experimental validation, emphasizes the potential of data-driven strategies for accelerating materials discovery. Future research will aim to broaden the dataset by incorporating diverse synthesis conditions and accounting for kinetic factors, thereby improving the robustness and generalizability of machine learning models for complex alloy systems.
4. Conclusion
-
Funding
This research is funded by the Ministry of Education and Training of Vietnam under grant number B2025-BKA-08.
-
Conflict of Interest
The authors have no conflicts of interest to declare.
-
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
-
Author Information and Contribution
Nguyen Lam Khoa: MSc student; responsible for conceptualization, experimental design and execution, original draft preparation.
Nguyen Duy Khanh: Undergraduate student; contributed to experimental work and performed calculations related to the mechanical alloying and Midema’s model.
Nguyen Thi Hoang Oanh: Associate Professor; supervised the research activities, provided critical revisions, and contributed to reviewing and editing the manuscript.
Le Hong Thang, Hoang Thi Ngoc Quyen: Ph.D Research members; contributed to conceptualization and conducted computational analyses supporting experimental findings.
Nguyen Hoa Khiem: MSc candidate; developed the Python code and user interface, and optimized the computational application for data analysis.
Nguyen Hoang Viet: Associate Professor; provided overall supervision, contributed to conceptual development, acquisition of funding, and participated in reviewing and editing the final manuscript.
-
Acknowledgments
None.
Article information







| Gradient Boosting | Random Forest | CatBoost | Experimental | |
|---|---|---|---|---|
| Al30Cu17.5Fe17.5Cr17.5Mn17.5 | BCC | BCC | BCC | BCC after MA and heat treatment |
| Cu20Cr20Fe20Mn20Ni20 | FCC | FCC | FCC | FCC |
- 1. J.-W. Yeh: JOM, 67 (2015) 2254–2261.ArticlePDF
- 2. W. Xiong, A.X.Y. Guo, C.T. Liu and S.C. Cao: J. Mater. Sci. Technol, 142 (2023) 196.Article
- 3. X. Wu: J. Mater. Sci. Technol, 147 (2023) 189.Article
- 4. T. G. Oliveira, D. V. Fagundes, P. Capellato, D. Sachs and A. A. A. P. Silva: Metals, 12 (2022) 1940.Article
- 5. J. Feng, Y. Tang, J. Liu, P. Zhang, C. Liu and L. Wang: Front. Bioeng. Biotechnol., 10 (2022) 977282.Article
- 6. X. Wang, W. Guo and Y. Fu: J. Mater. Chem. A, 9 (2021) 663.Article
- 7. F. Marques, M. Balcerzak, F. Winkelmann, G. Zepon and M. Felderhoff: Energy Environ. Sci., 14 (2021) 5191.Article
- 8. C. Han, Q. Fang, Y. Shi, S. B. Tor, C. K. Chua and K. Zhou: Adv. Mater., 32 (2020) 1903855.Article
- 9. D. B. Miracle: Nat. Commun., 10 (2019) 1805.Article
- 10. W. Zhang, P. K. Liaw and Y. Zhang: Sci. China Mater., 61 (2018) 2.ArticlePDF
- 11. Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang: Mater. Today, 19 (2016) 349.Article
- 12. M. H. Tsai and J. W. Yeh: Mater. Res. Lett., 2 (2014) 107.Article
- 13. B.S. Murty, J.-W. Yeh, S. Ranganathan: Chapter 6 – High-Entropy Alloy Solid Solutions, in High-Entropy Alloys, Elsevier (2014) 91.Article
- 14. B. Cantor: Entropy, 16 (2014) 4749.Article
- 15. J. W. Yeh: JOM, 65 (2013) 1759.ArticlePDF
- 16. J. M. Park: J. Powder Mater., 29 (2022) 132.Article
- 17. S. Lee, K. T. Kim, J.-H. Yu, H. S. Kim, J. W. Bae and J. M. Park: J. Powder Meter., 31 (2024) 8.ArticlePDF
- 18. W.-L. Hsu, C.-W. Tsai, A.-C. Yeh and J.-W. Yeh: Nat. Rev. Chem., 8 (2024) 471.ArticlePDF
- 19. C. Nagarjuna, S. K. Dewangan, H. Lee, E. Song, K. R. Rao and B. Ahn: J. Powder Mater., 32 (2025) 144.ArticlePDF
- 20. S. K. Dewangan, C. Nagarjuna, H. Lee, K. R. Rao, M. Mohan, R. Jain and B. Ahn: J. Powder Mater., 31 (2024) 480.ArticlePDF
- 21. S. V. Divinski, A. V. Pokoev, N. Esakkiraja and A. Paul: Diffus. Found., 17 (2018) 69.Article
- 22. D. B. Miracle: JOM, 69 (2017) 2130.ArticlePDF
- 23. R.K. Nutor, Q. Cao, X. Wang, D. Zhang, Y. Fang, Y. Zhang and J. Jiang: Adv. Eng. Mater., 22 (2020) 2000466.Article
- 24. R. F. Zhang, S. H. Zhang, Z. J. He, J. Jing and S. H. Sheng: Comput. Phys. Commun., 209 (2016) 58.Article
- 25. A. R. Miedema, P. F. de Châtel and F. R. de Boer: Physica B+C, 100 (1980) 1–28.Article
- 26. E. J. Pickering and N. G. Jones: Int. Mater. Rev., 61 (2016) 183.ArticlePDF
- 27. S. Singh, N. K. Katiyar, S. Goel and S. N. Joshi: Sci. Rep., 13 (2023) 4811.Article
- 28. R. S. Mishra, R. S. Haridas and P. Agrawal: Mater. Sci. Eng. A, 812 (2021) 141085.Article
- 29. T. Xie and J. C. Grossman: Phys. Rev. Lett., 120 (2018) 145301.Article
- 30. D. Dey, S. Das, A. Pal, S. Dey, C. K. Raul, P. Mandal, A. Chatterjee, S. Chatterjee and M. Ghosh: J. Alloys Metall. Syst., 9 (2025) 100144.Article
- 31. K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh: Nature, 559 (2018) 547.ArticlePDF
- 32. D. Dai, T. Xu, X. Wei, G. Ding, Y. Xu, J. Zhang and H. Zhang: Comput. Phys. Commun., 175 (2020) 109618.Article
- 33. J. Dąbrowa and M. Danielewski: Metals, 10 (2020) 347.Article
- 34. M. C. Troparevsky, J. R. Morris, P. R. C. Kent, A. R. Lupini and G. M. Stocks: Phys. Rev. X, 5 (2015) 011041.Article
- 35. A. Takeuchi and A. Inoue: Mater. Trans., 46 (2005) 2817.Article
- 36. S. Guo and C. T. Liu: Prog. Nat. Sci. Mater. Int., 21 (2011) 433.Article
- 37. D. Xue, P. V. Balachandran, J. Hogden, J. Theiler, D. Xue and T. Lookman: Nat. Commun., 7 (2016) 11241.Article
- 38. P. Mandal, A. Choudhury, A. Mallick and M. Ghosh: Met. Mater. Int., 29 (2022) 38.ArticlePDF
- 39. L. Breiman: Mach. Learn., 45 (2001) 5.Article
- 40. N. T. H. Oanh, D. N. Binh, H. T. N. Quyen, N. H. Viet and A. M. Jorge Junior: Metall. Mater. Trans. A, 56 (2025) 532.ArticlePDF
- 41. N. T. H. Oanh, D. T. An and N. H. Viet: Materials, 17 (2024) 5627.Article
- 42. D. N. Binh, N. T. H. Oanh and N. H. Viet: J. Non-Cryst. Solids, 583 (2022) 121478.Article
- 43. D. N. Binh, N. T. H. Oanh and N. H. Viet: Appl. Sci., 12 (2022) 16.Article
- 44. N. Q. Dung, L. V. Khoi, N. L. Khoa, V. Hai, D. X. Thanh, P. Anh, N. T. H. Oanh, N. D. Tuyen, N. M. Duc, L. H. Thang, P. H. Long, H. T. M. Loan, C. T. M. Thu, T. T. Phong, B. L. Hung, M. K. Luan, N. Q. Huy, V. Hiep, P. M. Tuan, N. M. Thuyet, N. H. Viet; Unveiling Phase Changes in Iron Ore Reduction, X-Ray Diffraction Analysis with a User-Friendly Colab Interface Integrated with the Materials Project API. In: in the International Conference on Advanced Materials and Technology (ICAMT 2024); (2024) p. 171.
- 45. T. Saito and M. Rehmsmeier: PLOS ONE, 10 (2015) e0118432.Article
- 46. B. Cantor: High Entropy Alloys Mater., 2 (2024) 277.ArticlePDF
- 47. M. Vaidya, G. M. Muralikrishna and B. S. Murty: J. Mater. Res., 34 (2019) 664.Article
- 48. P. Yu, L. Zhang, H. Cheng, H. Zhang, Y. Li, G. Li, P. Liaw and R. Liu: Intermetallics, 70 (2016) 82.Article
- 49. S. Joo, H. Kato, M. Jang, J. Moon, E.-B. Kim, S.-J. Hong and H.-S. Kim: J. Alloys Compd., 698 (2017) 591.Article
- 50. T. Lookman, P. V. Balachandran, D. Xue and R. Yuan: NPJ Comput. Mater., 5 (2019) 21.Article
- 51. D. Jha, L. Ward, A. Paul, W. K. Liao, A. Choudhary, C. Wolverton and A. Agrawal: Sci. Rep., 8 (2018) 17593.Article
References
Figure & Data
References
Citations
- Effect of annealing temperature on thermal expansion and cryogenic mechanical properties of low-thermal-expansion Co22.2Cr6.2Fe48.8Ni17.8Cu5.0 medium-entropy alloy
Wooyoung Lee, Munsu Choi, Sungwook Kim, Dae-Kyeom Kim, Myungsuk Song, Taek-Soo Kim, Jungwan Lee, Hyoung Seop Kim, Hyunjoo Choi, Soo-Hyun Joo
Materials Science and Engineering: A.2026; 954: 149811. CrossRef - Amorphization–Densification Coupling Governs Hardness Enhancement in SPS-Consolidated Al–Fe–Nb–(Ni,Ti) Metastable Alloys
Nguyen Thi Hoang Oanh, Nguyen Hoang Viet
Materials.2026; 19(12): 2628. CrossRef - Cr-Fe-Ni-Al designed from Cr-Fe-Co-Ni-Al high entropy alloys by ‘equal VEC strategy’ with significant reduction in cost and comparable mechanical properties
Chen Zhao, Ying Lin, Ziming Chen, Yangyang Cheng, Xinyu Li, Zhongyao Sun, Ruofei Deng, Yanzhe Wang, Chen Chen
Journal of Alloys and Compounds.2026; 1079: 190155. CrossRef - Preparation and Arc Erosion Behavior of Cu-Based Contact Materials Reinforced with High Entropy Particles CuCrNiCoFe
Jiacheng Tong, Jun Wang, Huimin Zhang, Haoran Liu, Youchang Sun, Zhiguo Li, Wenyi Zhang, Zhe Wang, Yanli Chang, Zhao Yuan, Henry Hu
Metallurgical and Materials Transactions B.2025; 56(5): 5948. CrossRef - Recent progresses on high entropy alloy development using machine learning: A review
Abhishek Kumar, Nilay Krishna Mukhopadhyay, Thakur Prasad Yadav
Computational Materials Today.2025; 8: 100038. CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article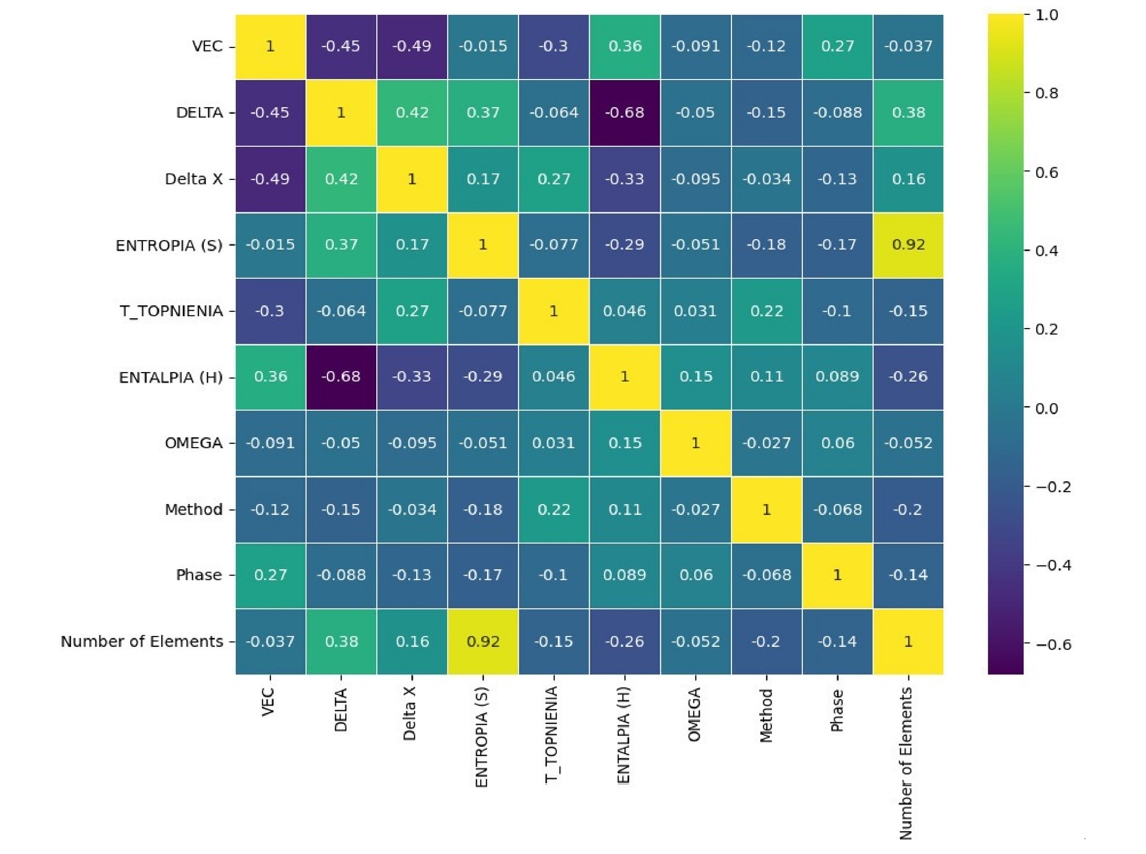
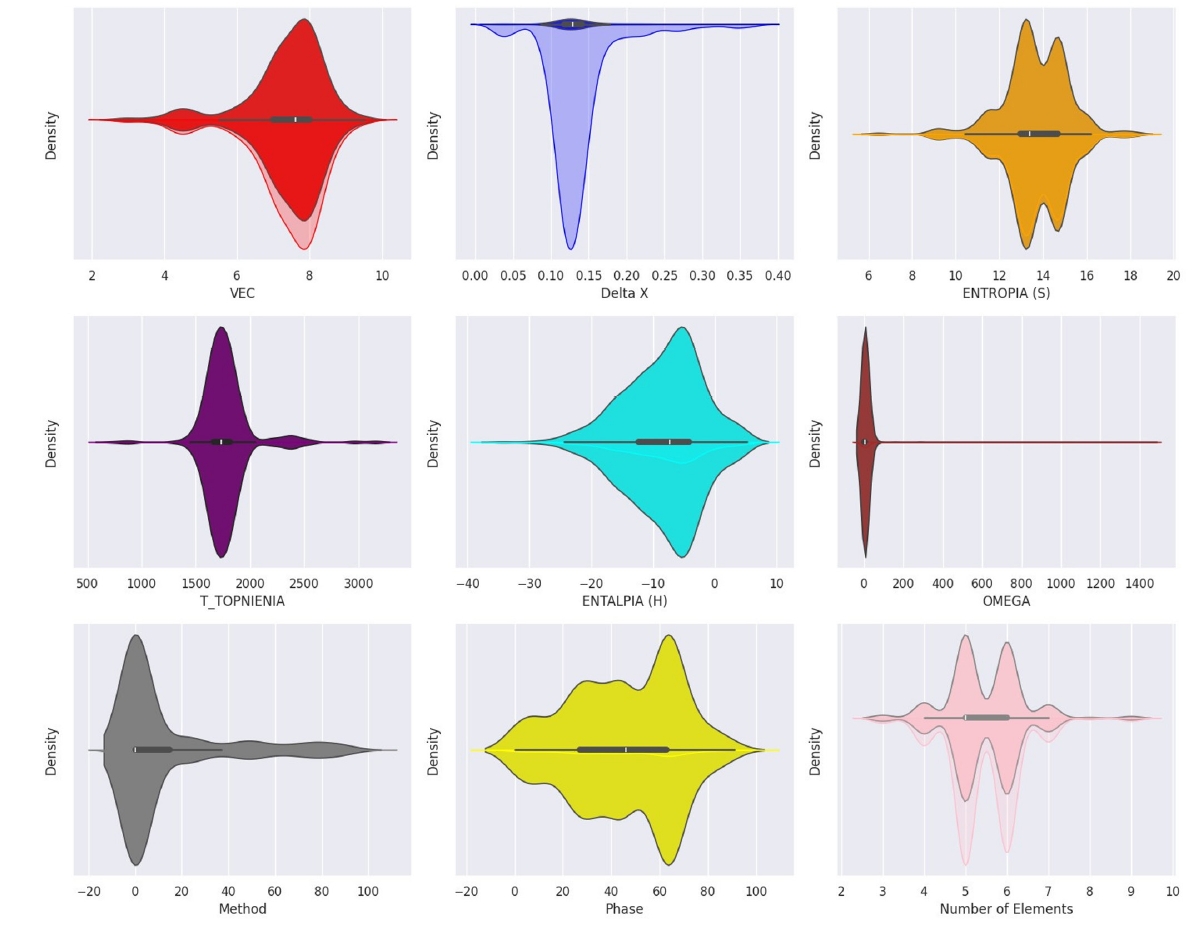
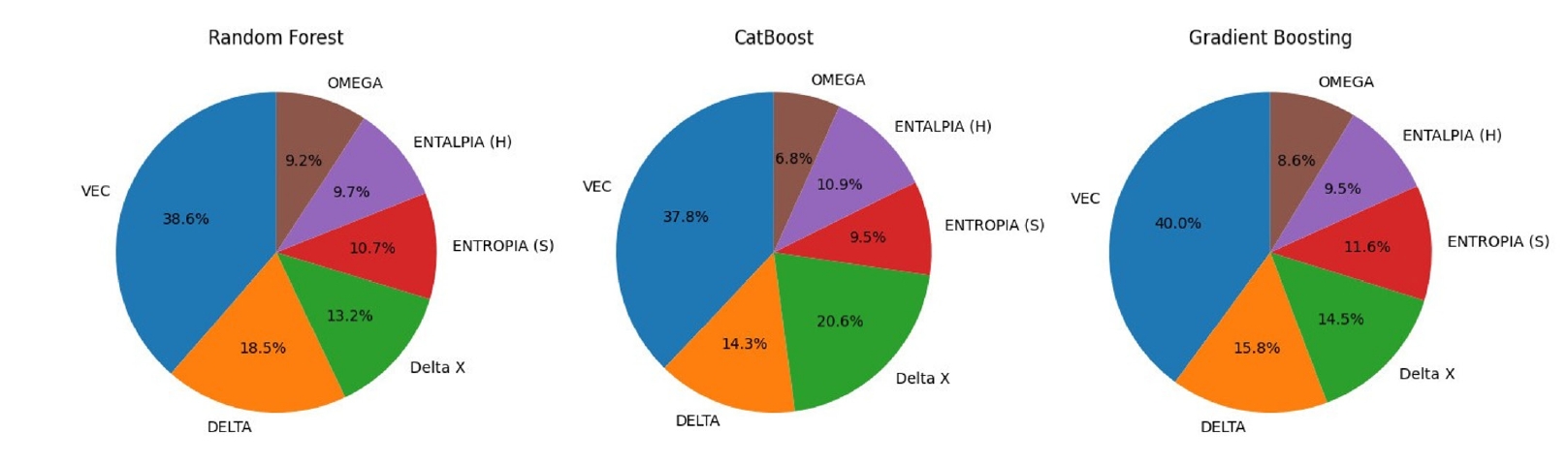
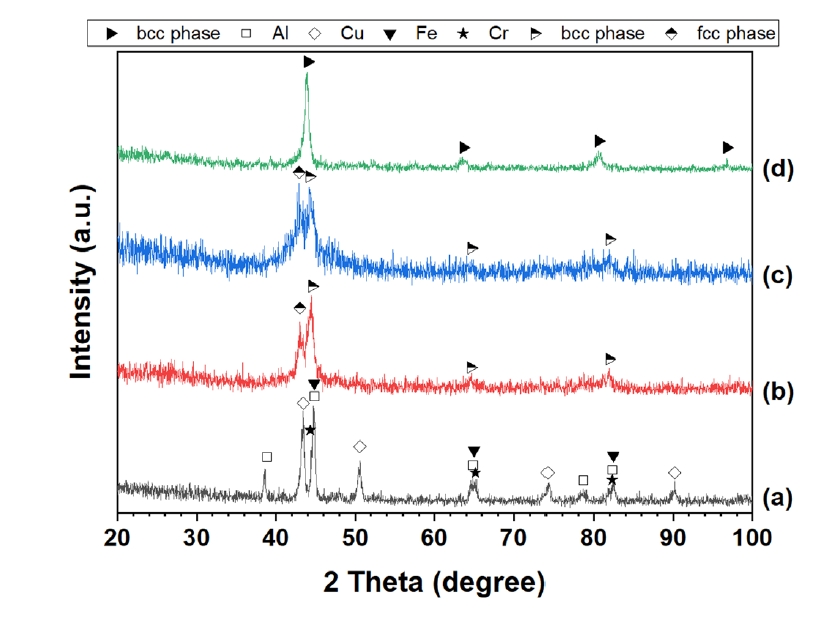
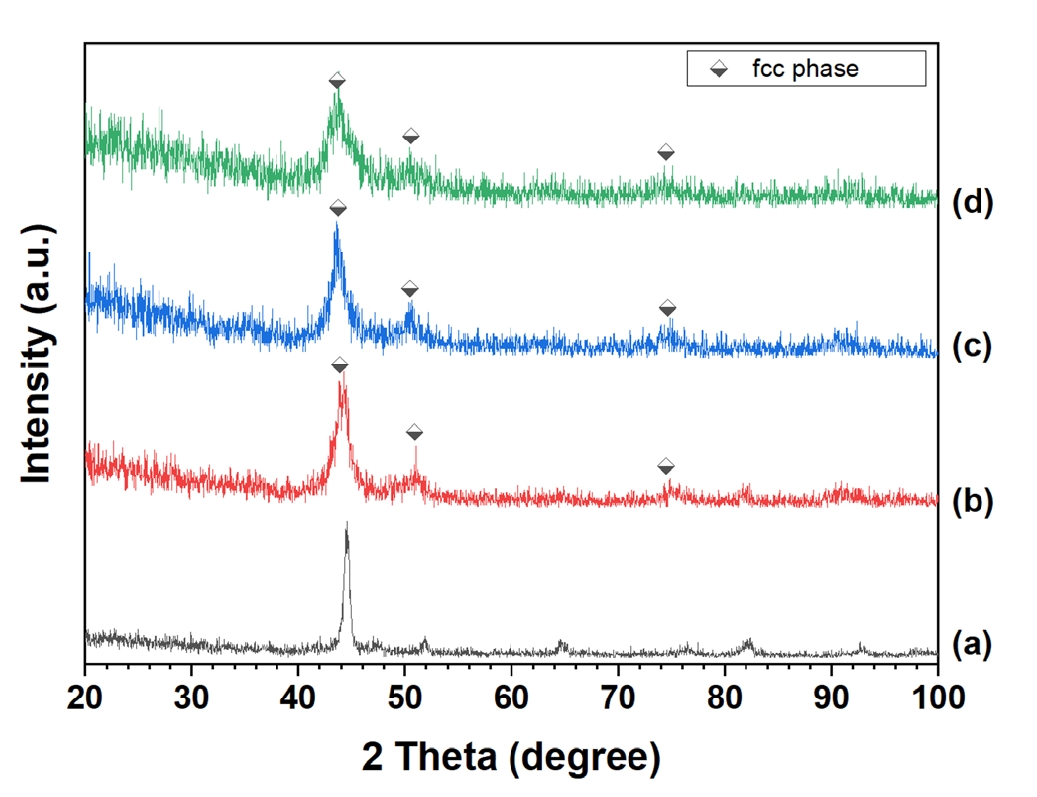

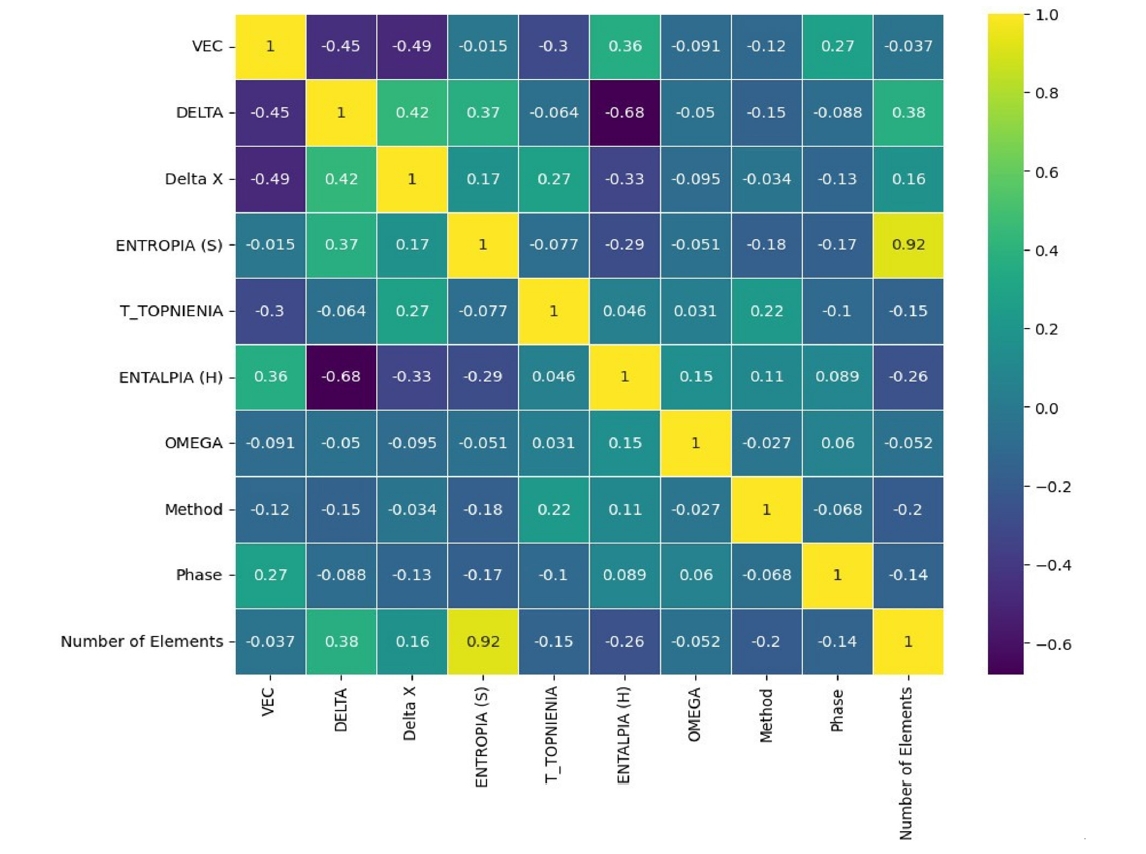
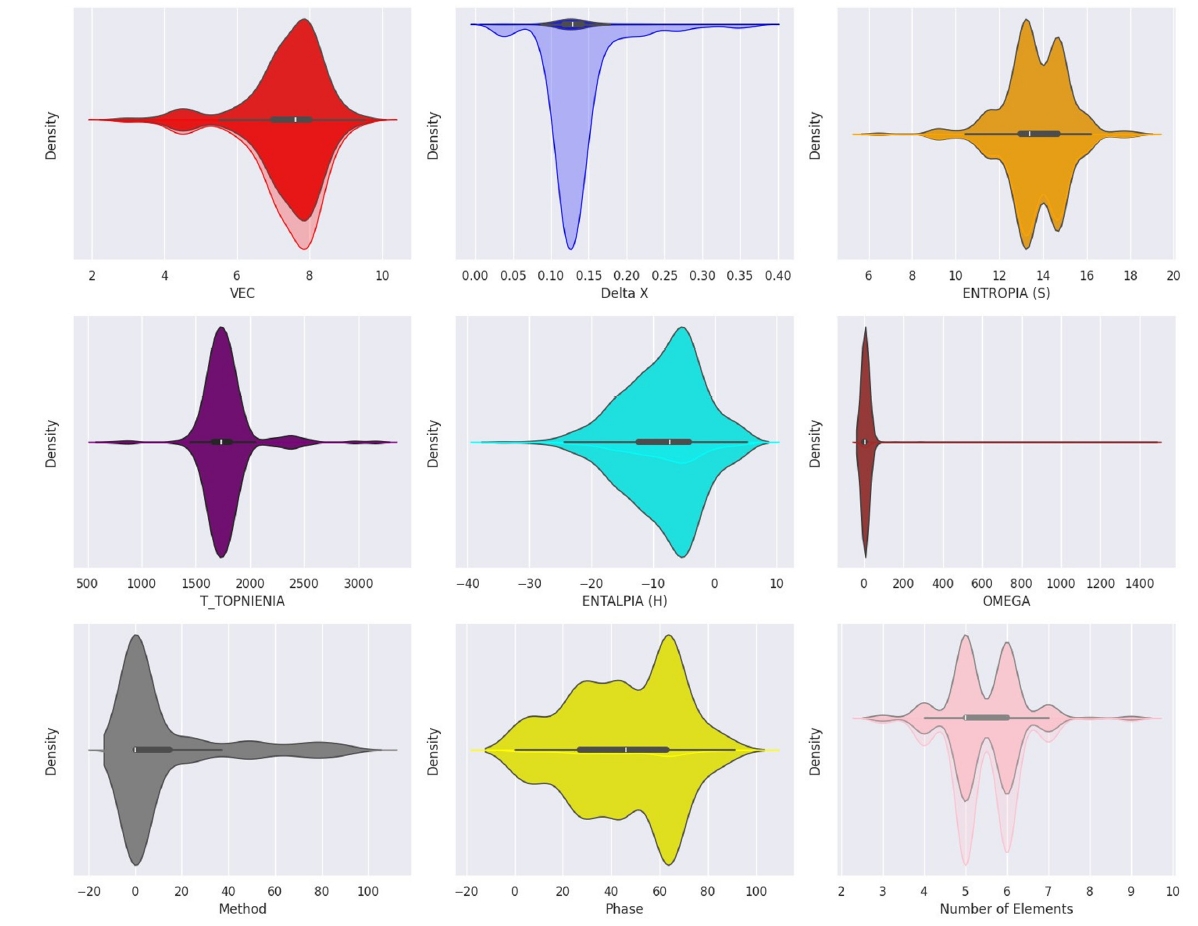




Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
Fig. 6.
Graphical abstract
| Mean | Std. Dev | Min | Max | |
|---|---|---|---|---|
| VEC | 7.363 | 1.072 | 2.800 | 9.500 |
| δ | 4.770 | 2.477 | 0.376 | 19.110 |
| Δχ | 0.136 | 0.045 | 0.031 | 0.364 |
| ΔSmix | 13.654 | 1.462 | 6.466 | 18.215 |
| ΔHmix | -8.335 | 6.316 | -34.348 | 5.125 |
| Tm | 1768.783 | 237.338 | 701.456 | 3155.500 |
| Ω | 8.589 | 46.134 | 0.836 | 1002.369 |
| Preset Range | Random Forest | Gradient Boosting | CatBoost | |
|---|---|---|---|---|
| max_depth (depth) | [1,9] | 7 | 5 | 8 |
| n_estimators (iterations) | [50,200] | 50 | 100 | 100 |
| min_samples_leaf | [1,5] | 2 | 2 | x |
| min_samples_split | [1,4] | 2 | 2 | x |
| l2_leaf_reg | [1,5] | x | x | 1 |
| learning_rate | [0.01, 0.1] | x | 0.07 | 0.1 |
| Best CV Score (Accuracy) | x | 0.833 | 0.885 | 0.846 |
| Gradient Boosting | Random Forest | CatBoost | Experimental | |
|---|---|---|---|---|
| Al30Cu17.5Fe17.5Cr17.5Mn17.5 | BCC | BCC | BCC | BCC after MA and heat treatment |
| Cu20Cr20Fe20Mn20Ni20 | FCC | FCC | FCC | FCC |
Note: x is “not applicable”
BCC, body-centered cubic; FCC, face-centered cubic.
Table 1.
Table 2.
Table 3.
TOP